



서론
1990년대 초, 일본의 SONY가 리튬이온 전지(Lithium-ion batteries, LIBs)를 최초로 상용화한 이후부터 현재에 이르기까지 LIBs는 휴대폰, 노트북 등의 소형화 전원에서부터 전기 자동차(Electric vehicles, EVs) 및 그리드 규모의 에너지 저장 시스템 (Energy storage systems, ESSs) 등의 대용량 전원까지 폭넓게 활용되어오고 있다. 특히, 최근 들어 LIBs가 탄소 중립 실현에 있어 중요한 역할을 하는 EVs에 성공적으로 도입이 되었고 동력원인 LIBs에 대해 더욱 높은 에너지 밀도, 장수명, 열적 안정성, 및 고속 충-방전 특성을 향상시키며 양적으로나 질적으로 괄목할 만한 성장을 이루어 내고 있다. 차세대 전지 개발에 있어 성능 향상 이외에도 추구해야할 하나의 요인으로 가격 경쟁력을 들 수 있는데, LIBs의 핵심 원료가 되는 리튬 화합물(수산화 리튬(LiOH)과 탄산 리튬(Li2 CO3))의 경우 한정된 리튬 자원 매장량과 특정 국가에 분포하는 지리적 편중성으로 인해, 상당한 가격 불안정성을 보인다. Fig. 1.a 에 나타나 있듯이, LIBs의 핵심 양극(cathode) 소재를 합성하는데 필요한 Li2 CO3의 kg 당 가격이 꾸준히 상승하고 있으며 시기에 따라 가격이 4배 이상 차이나는 등 향후 리튬 원료의 가격 변동성에 대한 우려[1]가 다수 존재한다. 수명이 다한 폐 LIBs로부터 양극재 원료의 재활용 공정을 거쳐 리튬 원료를 수급하는 산업 기술들이 개발되어지고 있으나, 앞으로 성장될 LIBs의 시장규모로 미루어 볼 때, 보다 안정적인 리튬 원료 수급처를 마련하는 것은 필수적인 산업 과제라 할 수 있다.
Fig. 1.
a. Historical and prospective prices of essential metals (cobalt, manganese, nickel, and lithium) applied in battery technologies. Reprinted with permission.[1] Copyright 2024, Elsevier. b. Mass fraction of elements in the Earth's crust. Reprinted with permission.[2] Copyright 2018, WILEY-VCH Verlag GmbH & Co. KGaA
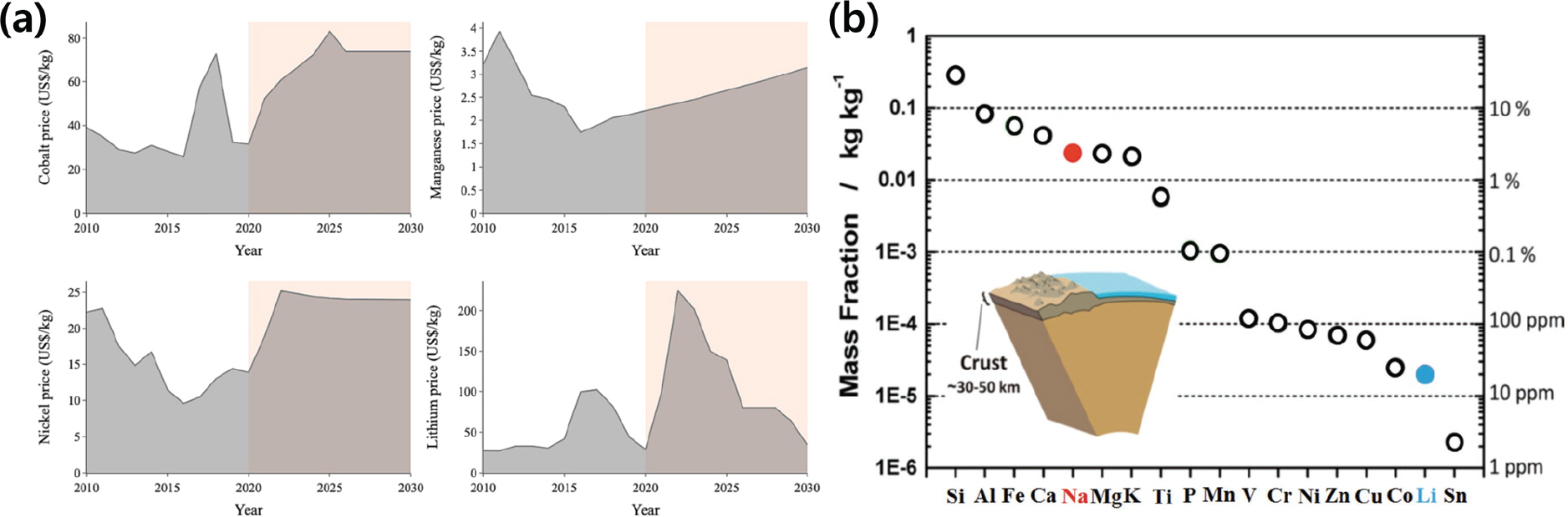
최근, 이러한 리튬의 가격 불안정성으로 인해 차세대 전지 시스템으로 소듐 이온 전지(Sodium-ion batteries, SIBs)가 학계 및 산업계에서 큰 주목을 받고 있다. 소듐의 경우 Fig. 1.b 에 나타난 바와 같이, 지구상에 5번째로 풍부한 원소로 원재료 가격이 리튬에 비해 훨씬 저렴하며 지각의 암염(rock salt)이나 해수의 염(salt)으로 존재하여 지역 편중성이나 고갈될 위험이 없다.[2] 또한, 소듐은 리튬과 같은 알칼리 금속 족 내 가장 인접한 원소로써 이온 반경, 무게, 그리고 표준 환원 전위 (standard reduction potential)가 다른 원소들에 비해 상대적으로 유사하기 때문에(Table 1) 고에너지밀도의 전지 구현이 가능하다.[3,4] 또한, 기존의 LIBs 시스템에서는 음극의 반응전압 영역에서 Li-Al 합금상이 형성되는 문제로 인해 집전체(current collector)로 값싼 알루미늄(Aluminum) 대신 비싼 구리(Copper)를 사용하게 되는데 SIBs 시스템에서는 Na-Al 합금상 형성에 대한 문제가 없어 음극 집전체로 값싼 알루미늄을 사용할 수 있다는[5] 추가적인 장점도 있다. 이러한 장점들로 인해서 현재 국내/외 이차전지 유관 산업체 및 연구소들이 SIBs 를 연구 개발하는 중에 있고, 특히 중국의 CATL 기업이 SIBs의 상용화를 근 시일 내 추진하겠다고 발표한 바 있다.
Table 1.
Comparison between lithium(Li) and sodium(Na). Reprinted with permission.[4] Copyright 2013, Wiley-VCH Verlag GmbH & Co. KGaA
| 범주 | 리튬 (Li) | 소듐 (Na) |
|---|---|---|
| 원자량 (g mol-1) | 6.94 | 22.99 |
| 1가 양이온 반경 (Å) | 0.76 | 1.02 |
| 표준 환원 전위 (V vs. SHE) | -3.04 | -2.71 |
| 융점 (° C) | 180.5 | 97.7 |
| 이온화 에너지 (kJ mol-1) | 520.2 | 495.8 |
| 지각 내 매장량 (mg kg-1) | 20 | 23.6×103 |
| 주요 분포 | 남미, 중국, 호주 | 광범위 |
| 탄산염 가격 ($ ton-1)[3] | 37,000 | 140 |
| 무게당 이론 용량 (mAh g-1) | 3861 | 1161 |
| 체적당 이론 용량 (mAh cm-3) | 2062 | 1131 |
하지만, 경제적 측면에서 장점을 가지는 SIBs를 상용화하는데 극복해야할 몇 가지 문제점들이 존재하게 되는데, 먼저 알칼리 금속 중 소듐은 리튬 다음으로 이온 크기가 작고 가벼우며 리튬과 비슷한 표준 환원 전위를 가졌음에도, 리튬에 비해서는 이온이 크고 무거우며, 높은 환원 전위를 가지기 때문에 에너지 밀도가 더 낮을 수밖에 없다. 또한, 무겁고 큰 소듐 이온의 느린 확산으로 인해 충-방전 시 큰 저항의 증가를 야기하게 되며, 이온 저장/사용에 따른 전극 부피변화가 커 전극의 구조적 안정성이 떨어지게 된다. 무엇보다도, 다음 장에서 소개할 기존의 LIBs용 상용 전극소재들(흑연, 실리콘 등)이 SIBs 시스템에 잘 적용되지 않는다는 점이 소재 연구 관점에서 해결해야할 최우선 과제이다. 따라서, 본 리뷰 논문에서는 기존에 LIBs에서 상용화 되어 있거나, 상용화를 추진 중인 음극 소재들이 SIBs 용 음극으로 사용되었을 때의 문제점과 기존 소재들을 대체하기 위한 SIBs 용 음극 소재 개발 연구 동향에 대해 소개하고자 한다.
본론
2.1 흑연 기반 음극 소재 연구
흑연(Graphite)은 탄소(Carbon) 소재의 여러 동소체 중 하나로, 순수한 탄소 원자 1개가 3개의 다른 탄소 원자와 강한 공유결합을 형성하여 sp2 혼성 궤도(sp2 hybridization orbital)로 이루어진 평면 육각형 격자 형태로 잘 알려져 있다(Fig. 2.a). 일반적으로, 육방정계(hexagonal) 흑연의 각 층은 2차원의 그래핀(graphene) 격자들이 c축 방향으로 ABABAB 적층순(stacking sequence)으로 적층 되어 있고, 각 그래핀 격자층 간의 파이(π)-결합은 약한 반 데르 발스(Van der waals) 결합으로 이루어져 있다.[6] 이러한 결정 구조를 가지는 흑연의 층간 사이에 리튬 이온이 삽입(intercalation) 및 탈리(de-intercalation)가 가능함으로써 양극과 음극 사이를 오가는 셔틀(shuttle) 이온인 리튬 이온의 저장과 방출이 가능하다. 이처럼, 실제 상용화 된 LIBs에서는 외부에서 전압을 인가해주는 충전 반응(charging reaction)을 통해 리튬 이온이 흑연의 층상 구조 내로 삽입되게 되는데, 이는 층상 구조의 양극 재료(e.g., LiCoO2(LCO) 또는 LiNi1-x-y Co x Mn y O2(NCM))에서 산화 반응을 통해 빠져나온 리튬 이온과 전자가 흑연에서 xLi+ + xe- + C6 ↔ Li x C6 의 환원 반응을 통해 Li x C6 화합물을 형성하게 된다. 리튬 이온은 흑연의 층간 구조 내에 무작위로 삽입 및 탈리되는 것이 아니라 특정한 규칙성을 가지는데 이를 스테이징(staging) 현상이라고하며, R R dor RR-Ho R mann 모델과 Daumas-H´ erold 모델 등(Fig. 2.b)이 제시되어 있다.[7-11] 흑연의 층 간에는 다양한 분자나 원자를 삽입할 수 있으며, 그 물질에 따라서 전기 전도성이 결정되게 된다. 금속 이온을 삽입하는 경우, 금속의 자유전자가 흑연으로 이동하여 흑연 층 내부에서 움직이며 그래핀 층 내 평면 방향 전기 전도도가 증가하는 특징이 있으며, 리튬이 삽입된 흑연 역시 전도성이 잘 유지되는 동시에, 층간 구조에 리튬 이온 삽입/탈리 시 부피변화가 거의 없어 안정적인 전극 구조를 유지하여 LIBs 상용화에 주요한 역할을 해오고 있다.
Fig. 2.
a. Crystal structure of graphite. b. Schematic illustration of staging structures of lithiated graphite with different models. Reprinted with permission.[10] Copyright 2022, Wiley-VCH Verlag GmbH & Co. KGaA

이와 같이, LIBs의 상용화를 앞당긴 안정적이고 값싼 흑연 소재를 SIBs 음극 소재로 활용하고자 하는 시도가 있다. SIBs용 흑연의 초기 연구 결과에 의하면, 소듐 이온과 흑연의 전기화학 반응 시, 약 35 mAh g-1 의 매우 낮은 초기 가역 용량을 보이며 가역적으로 삽입/탈리 반응이 NaC186 화합물을 형성하는 것으로 계산되는 등 소듐 이온이 흑연층에 미미하게 삽입되는 것으로 보고되었다.[8,9] 이 같은 낮은 삽입 반응성의 이유로, 이원계 흑연 삽입 화합물(Binary graphite intercalation compound)을 형성하기 위해 소듐 이온이 삽입될 때 그래핀 층과의 상호작용에서 있어 열역학적 안정성이 다른 리튬 또는 포타슘 이온에 비해 불리하다는 DFT 이론(Density functional theory) 기반 컴퓨터 시뮬레이션 결과가 제시된 바 있다.[12,13]
최근 연구에 따르면, 소듐 이온이 전해질의 용매와 반응하여 용매화된 분자사슬의 형태로 흑연의 층 간 간격 사이로 공동 삽입(co-intercalation)될 수 있고 이를 통해 삼원계 흑연 삽입 화합물(Ternary graphite intercalation compound)이 형성된다는 것이 밝혀졌다.[13-15] 소듐 이온, 용매(diethylene glycol dimethyl ether, G2), 그리고 흑연을 포함하는 삼원계 화합물의 공동 삽입 반응은 [Na− G2]+로 용매화된 소듐 이온이 흑연 층간 구조에 삽입한다.[14] 이러한 공동 삽입 반응은 율속 반응 속도 (rate capability) 시험에서도 뛰어난 전기화학적 성능을 보여주었고 전류밀도 100 mA g-1 기준 약 110 mAh g-1의 향상된 가역 용량 거동이 확인되었다. 이처럼, 소듐 이온의 흑연 층간 구조 삽입에 관하여 공동 삽입 방식, 소듐 염(salt) 및 용매 조건 변화 등 다양한 시도가 이루어지며 가역 용량이 향상되고는 있으나, 기존 흑연이 LIBs에서 보이는 이론용량인 372 mAh g-1(LiC6 상 형성 시)에는 크게 못 미치는 상황으로 SIBs의 낮은 가역 용량 향상이 필요한 상황이다.[16,17] 용량 측면 뿐만 아니라, 현재 개발된 흑연 내 용매화된 소듐 이온의 공동 삽입/탈리 반응 전위가 0.6∼0.8 V (vs. Na/Na+) 수준으로 LIBs에서 보이는 0.15 V (vs. Li/Li+) 근처 전위에 비해 높아 에너지 밀도가 낮기 때문에 반응 전위 조절의 문제도 고려되어야 할 연구 방향 중 한가지라 할 수 있겠다.
2.2 실리콘 기반 음극 소재 연구
상기 소개된 흑연 소재의 SIBs 적용에 대한 연구 흐름 이후, LIBs에서 가장 높은 이론 용량을 보이는 고용량 실리콘(Silicon, Si) 소재를 SIBs에 적용하는 연구가 진행되어졌다.[18] 실리콘은 지구상 지각 매장량이 가장 풍부하고(Fig. 1.b) 독성이 없으며 리튬 이온과 합금 반응(alloy reaction, 4.4 Li+ + 4.4 e- + Si ↔ Li4.4 S i) 시 실리콘 원자 1개 당 약 4.4개의 리튬 이온과 반응하여 약 4200 mAh g-1의 이론 용량을 보인다.[19] 이런 고용량 소재를 음극재로 활용하게 되면 전극 내 필요한 전체 전하(charge)를 저장하기 위해 요구되는 음극재의 부피나 무게를 줄일 수 있어 고에너지 밀도의 전지를 구성할 수 있다. 하지만, 다르게 해석하면 고용량 음극 소재는 하나의 호스트 원자(host atom)에 저장되는 리튬 이온의 수가 많아지기 때문에 많은 이온의 출입에 따른 큰 부피 팽창/수축이 발생하게 된다.[19-21] 이는 전극 단위에서 음극재 입자의 박리와 단위 입자에 균열 및 파쇄를 야기하고, 고체-전해질 계면(solid electrolyte interphase, SEI) 층의 부반응(side reaction)을 증가시켜 비가역적인 용량(irreversible capacity)을 증가시키는 문제도 있다.[22,23] 이 때문에, 음극재의 부피 변화가 작은 흑연 소재에 부피 변화가 큰 실리콘 음극 소재를 소량 첨가하여 흑연/실리콘 복합체(composite)를 형성하여 전극 부피 변화의 문제를 최소화하여 상용화하고자 하는 노력이 이어지고 있다.[24,25]
이와 같은 배경을 토대로 고용량 실리콘 음극을 SIBs 에 적용하고자 연구가 진행되고 있는데 소듐-실리콘의 2성분계 상태도(binary phase diagram)를 참고하면 소듐-실리콘 화합물에서 NaSi 조성이 가장 소듐 분율이 높은 상으로 알려져 있다.[26-28] 이는, SIBs에서 실리콘 원자 하나당 소듐 원자 하나와 반응하여 나타내는 이론용량이 954 mAh g-1 수준으로, 실리콘 원자 하나당 리튬 원자 4.4개와 반응하여 보이는 LIBs의 이론용량 4200 mAh g-1에 비해 약 23% 수준의 낮은 용량 특성이다.[27,29] 또한, 실제 마이크로 크기를 가진 c-Si(crystalline Silicon)을 SIBs 용 음극재로 연구 결과에 따르면, 가역 용량이 7.5 mAh g-1 (전류밀도: 36 mA g-1)수준으로 매우 낮게 나오며 탈소듐화의 반응이 일어나지 않아 큰 비가역성을 나타냈고, 나노 크기의 c-Si일 경우에는 방전 곡선에서 0.5 V 이하로 전위 평탄 구간을 보이며 약 1000 mAh g-1의 용량을 보였으나 가역 용량은 약 200 mAh g-1 (전류밀도: 500 mA g-1)수준으로 매우 저조한 특성을 나타났다.[30,31] 이처럼 SIBs에 실리콘 음극을 사용하게 될 경우, 이론적으로도 고용량 특성을 기대하기 힘들고, 합금/탈합금 반응 시 발생하는 큰 분극 등으로 인해 kinetic 측면에서 극복해야 할 과제가 많은 상황이다.
2.3 하드 카본 기반 음극 소재 연구
위 2.1 및 2.2 절에서 소개된 흑연과 실리콘 음극 소재 모두 SIBs에 적용되는데 있어 여러가지 문제점이 확인되었지만, 다른 탄소 계열 소재 중 하나인 하드 카본(Hard Carbon)의 경우 SIBs에 적용 시 LIBs에서의 특성과 상대적으로 유사하여 최근까지 많은 연구 개발이 지속되었다.[32,33] 최초 포도당 유래의 보고로 하여금 하드 카본의 가역 용량이 약 300 mAh g-1 임이 보고되었으며, 이후 섬유소, 설탕 전구체, 식물 기반 등 바이오 매스(biomass)를 통한 하드 카본의 연구가 시도되어왔다.[11,34,35] 초기 연구 결과에서는 하드 카본의 가역 용량 특성이 약 280-300 mAh g-1 (전류밀도: 35 μ A cm-2)으로 흑연 음극과 비교하여 우수한 성능을 나타내, 하드 카본 소재의 SIBs 응용 가능성이 큰 관심과 집중을 받았다.[34] 하드 카본은 3000° C에서도 열처리에 의한 흑연화가 되지 않는 특징이 있으며, 소듐 이온이 용매화되어 흑연 층간 구조 내에 공동 삽입되는 흑연의 반응 메커니즘과 달리, 소듐 이온과 하드 카본 사이의 명확한 반응 메커니즘에 대해서는 여전히 불분명한 부분이 있다. 하드 카본은 소듐 이온이 곧바로 삽입 및 흡착될 수 있는 무질서한 배열(turbostratic structure)의 유사 흑연 도메인(pseudo graphitic domains)을 가지고 있다.[36,37] 또, 소듐 이온이 확산되기 위한 경로를 제공하는 일부 다공성, 계층적 기공 크기, 표면 결함과 소량의 이종 원소(N, S, P등)도 불순물처럼 구조 내에 존재하고 있어 더 많은 소듐 저장 활성화를 촉진한다.[38] 하드 카본 내 소듐 이온의 저장 거동에 대해 크게 3가지 형태가 보고되고 있는데, i) 유사 흑연 도메인 내로의 소듐 이온 삽입(insertion), (ii) 표면 가장자리(edge), 결함(defect) 및 이종 원소에 대한 이온 흡착(absorption), (iii) 나노 기공 채움(nanopore filling)으로 분류된다 (Fig. 3.a).[36,38,39] 이러한 이온 저장 메커니즘은, 복합적으로 발생할 수 있고 하드 카본의 구조적 차이에서 달라질 수 있으며 주로 전구체 종류와 탄화 공정 조건에 크게 지배를 받는다. 그로 인해, 여전히 의문이 제기되는 영역은 상호 층 간 간격 및 나노 기공 주위에서 발생하는 이온 저장 과정이며, 이에 대한 메커니즘적 이해 연구가 진행되고 있다.[37,40]
Fig. 3.
a. Schematic illustration of the microstructure and the ion-storage sites of graphite and hard carbon. Reproduced with permission.[13,36] Copyright 2021, American Chemical Society. b. Rate capabilities of the hard carbon electrodes with different carbonized temperature of 1000, 1300 and 1600 o C, respectively. Reprinted with permission.[35] Copyright 2016, Wiley-VCH Verlag GmbH & Co. KGaA

현재 하드 카본이 SIBs용 고성능 음극 소재로써 활용되기 위해 넘어야할 기술적 장벽은 0.1 V vs. Na/Na+ 부근의 낮은 반응 전압으로 인해 발생하는 초기 비가역적 용량과 저조한 율속 특성(rate capability)이다.[41] 탄화공정 온도(carbonization process temperature)에 따른 하드 카본의 SIBs용 음극 특성 연구를 참고하면, 탄화 온도 1300 o C로 최적화된 하드 카본의 경우 0.1 C(=30 mA g-1) 전류밀도에서 약 310 mAh g-1의 가역용량을 나타내는데 반해, C-rate이 1C, 2C로 증가하는 경우 각 200과 100 mAh g-1으로 가역용량이 크게 감소되는 열악한 율속 특성을 나타내었다(Fig. 3.b). [35] 이러한 단점들을 해결하기 위해 다양한 전략들이 연구되고 있으며 주로 불균일 원소 도핑, 나노 복합체화, 높은 탄화 공정 온도, 화학적-물리적 활성 변화를 위한 기공율 변화, 화학적 소듐화 전처리, 결함 관련 공학 등 하드 카본의 전처리와 후처리 공정 연구가 이루어지고 있다.[37]
2.4 금속 산화물 기반 음극 소재 연구
위 2.1 및 2.3절에서 다룬 흑연과 하드카본 소재의 경우 이온 저장 메커니즘이 삽입/탈리 반응에 기초하고 있어 충-방전 반응 시 소재의 부피 변화가 적기 때문에 안정적인 수명 특성을 보이는 소재들이다. 각 소재들의 상기 서술된 문제들로 인해 이온 저장 메커니즘이 삽입/탈리인 다른 음극 소재들에 대한 연구 개발이 이루어지고 있는데 대표적인 물질군으로 전이 금속 산화물 소재가 있다. 다양한 전이금속 산화물 중 특히, 4-6족 사이의 초기 전이금속(early transition metal)을 포함하는 산화물의 경우 격자 구조 내 초기 전이금속 양이온의 크기가 충분히 커서 타 이온을 삽입할 공간이 크고, 양이온의 전기음성도가 충분히 작아 Li/Li+ 또는 Na/Na+ 환원 전위 근처에서도 환원되지 않고 구조 내 리튬 또는 소듐 이온이 삽입/탈리 될 수 있는 구조를 유지하는 삽입/탈리 반응 소재가 많이 존재한다. 전이 금속 산화물 내 대표적인 삽입/탈리 반응 음극 소재는 타이타늄 산화물(TiO)과 바나듐 산화물(V-O)을 들 수 있다.[42,43]
2.4.1 타이타늄 산화물계 음극 소재
타이타늄 산화물의 경우 초기 전이금속 중 가장 가벼운 타이타늄 원소로 구성되어 있기 때문에 음극 소재 무게 측면에서 더 높은 무게 대비 용량 (specific capacity)을 나타낼 수 있는 물질이다. 특히, Fig. 4.a 와 같이 여러 다형성(polymorphs)[44]을 지닌 TiO2(titanium dioxide) 중 Anatase 구조의 TiO2가 3차원 개방 구조를 통해 리튬 이온 삽입과 확산에 가장 이점이 있는 소재로 알려져 있으며, 이는 고출력, 급속 충전을 실현시키는 음극 소재로 기대를 받아왔다. Li7 Ti5 O12(LTO) 음극 또한 TiO2와 유사한 구조적 특성으로 셔틀 이온의 출입이 빠르고 가역적이어서 고출력, 장수명 등 특성이 뛰어나다는 장점이 있다.[45] 이외에도 화학적 안정성, 비독성, 경제성 등의 장점을 토대로 고출력/급속 충전이 가능한 SIBs용 음극으로 타이타늄 산화물계(Li7 Ti5 O12, TiO2)에 대한 기초적 연구가 시작되었다. LTO 소재는 LIBs 음극으로 적용 시 이론 용량이 175 mAh g-1로 흑연의 이론 용량(372 mAh g-1)보다 낮고 작동 전압이 약 1.5 V vs. Li/Li+로 높아 에너지 밀도 측면에서 큰 장점이 없던 소재이지만, 현재 흑연 및 하드 카본이 SIBs 용 음극에서 보이는 용량 수치가 뛰어나지 않기 때문에 연구 대상이 될 수 있었던 소재이다.[46] LTO와 유사한 조성을 가지는 Na2 Ti6 O13(NTO) 소재가 Wu 연구팀에 의해 연구되었는데, 초기 반응 시 소듐 이온이 NTO에 충분히 삽입되지 못하여 초기 가역 용량이 20 mA g-1의 전류 밀도에서 약 60 mAh g-1로 낮게 나타났으며, 산화/환원 전위가 약 1.0 V vs. Na/Na+에 가깝게 높았다. 50회 충-방전 싸이클 이후 용량 활성화 과정(capacity activation process)을 통해 가역 용량이 160.7 mAh g-1로 증가하는 추세를 보였으나, 80회 싸이클부터 일부 Ti+4 + 4e- → Ti0로 금속 상태로의 비가역적 환원과 NTO의 구조적 재구성에 의해 용량 손실이 관찰되었다.[47] 후속 연구로 형성된 터널형 구조의 NTO와 층상구조형 Na2 Ti3 O7의 복합체(composite)는 열악한 초기 용량 및 용량 활성화 현상 문제가 개선되었으나 고성능 음극 소재로 적용되기에는 초기 방전 용량이 약 120 mAh g-1로 낮은 형태를 보였다.[48]
Fig. 4.
a. Crystal structures of three TiO2 polymorphs (Rutile, Brookite, and Anatase). Reprinted with permission.[44] Copyright 2017, Springer Nature. b. Galvanostatic discharge/charge voltage profiles of amorphous TiO2 nanotube with an inner diameter of 80 nm at a current density of 50 mA g-1 (C/3), Reprinted with permission.[50] Copyright 2011, American Chemical Society
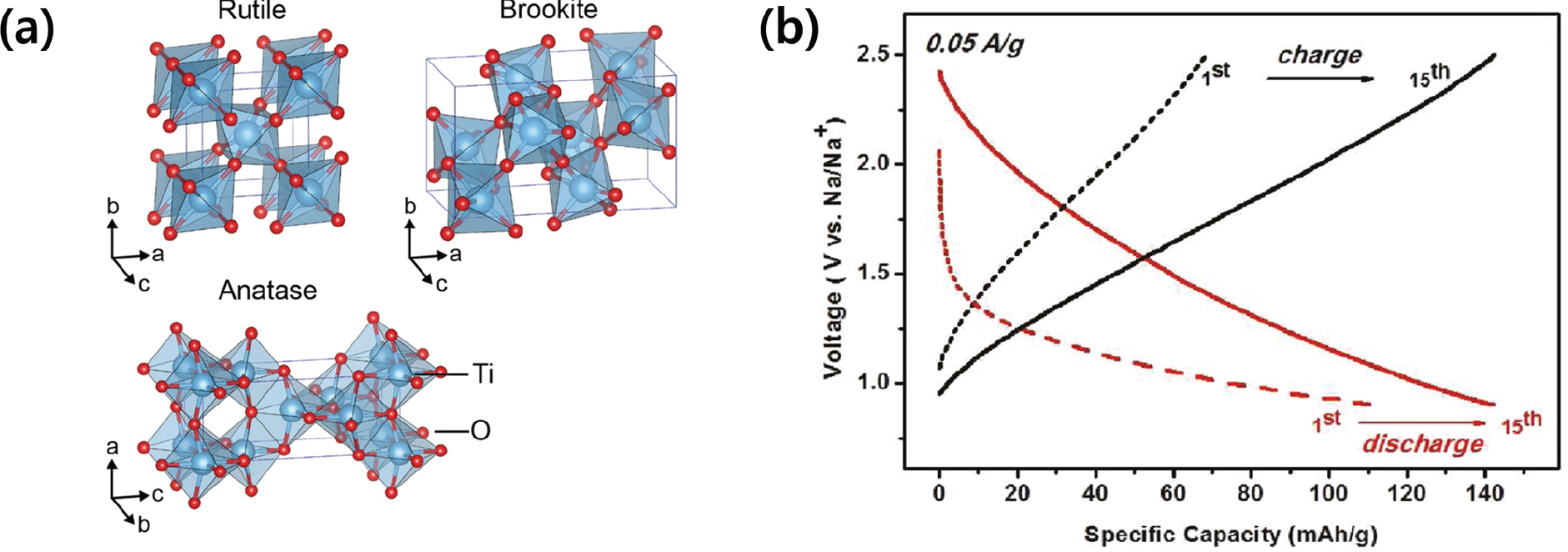
TiO2 구조의 경우 상기 언급된 LTO의 낮은 이온 저장 능력을 보완하기 위해 개발된 음극 소재로써 총 8개의 다양한 동질 이상 구조를 가져 광촉매, 이차전지, 태양 전지 등 첨단 산업에 응용되고 있는 재료이다.[49] 이러한 TiO2계 음극은 Ti4+/Ti3+의 산화/환원 반응으로 이온을 저장하는 방식으로 작동한다. 초기 Xiong 연구팀이 개발한 TiO2는 SIBs용 음극 소재로 활용하기 위해 비정질 1차원 TiO2 나노튜브로 합성되었고, 50 mA g-1에서 약 150 mAh g-1의 가역 용량을 나타낸 것으로 보고가 된 바가 있다(Fig. 4.b). 이는 나노 구조화를 통한 TiO2가 우수한 이온 확산도를 제공하고 높은 전기화학적 가역성을 나타낼 뿐더러 수직으로 정렬된 비정질 구조로 인한 효율적인 전기 전도성을 통해 전도성 탄소 첨가재나 바인더 등의 필요성을 제거하였다고 밝혔다.[49,50] 이외에도 다른 나노 구조 및 동질다형의 anatase-TiO2, rutile-TiO2, brookite-TiO2, bronze-TiO2(Fig. 4.a) 등에 대한 SIBs용 음극 소재들이 연구되었으나, anatase-TiO2가 소듐 저장을 위한 호스트(host)로 가장 적합하다는 연구 결과가 다수로 보여진다.[51-53] Anatase-TiO2의 초기 연구 과정에서는 리튬 및 소듐 이온의 삽입/탈리 과정 내 활성화 장벽(activation barrier)이 유사하다고 판단되었으나, 실제 anatase-TiO2의 LIBs 내 삽입/탈리 과정 중 특징적인 전위 평탄 구간이 보이는 것과 달리, SIBs 내에서는 전위 평탄 구간이 보이지 않는 것으로 나타난다. 이는 리튬 및 소듐의 삽입/탈리 전기화학적 반응에 명확한 차이를 나타내는 것으로 명확한 메커니즘의 차이는 아직 논의 중에 있다. 여러 가능한 에너지 저장 메커니즘이 제안되었는데, 삽입 반응(intercalation reaction), 전환 반응(conversion reaction) 그리고 유사 용량 저장(pseudocapacitive storage)이 포함된다.[54] anatase-TiO2의 격자 내로 소듐 이온이 삽입되는 삽입 반응 메커니즘은 소듐화 과정 동안 Ti4+/Ti3+의 산화수 변화(산화/환원 반응) 시 관찰되는 층 간 간격의 변화 및 그에 따른 회절 피크의 이동으로 제안된 반응 메커니즘이다. 또, 다르게 제안된 메커니즘은 anatase-TiO2의 소듐화 중에 형성되는 다양한 소듐 티타네이트 상(sodium titanate phase)과 티타늄 금속(titanium metal)으로의 상 전환을 하는 전환 반응 메커니즘이며, 다른 하나는 표면 반응(surface reaction)으로 TiO2의 소듐화, 탈소듐화 시 유사 용량 저장뿐만 아니라 표면에서 일어나는 Ti4+/Ti3+ 산화/환원 반응이 용량에 기여한다고 제안된 바 있다.[54] 이러한 메커니즘들의 불일치는 anatase-TiO2의 입자 크기, 결정성, 이온/전기 전도성, 충-방전 과정 중 국소적인 구조 변화 및 작동 조건 차이 등 종합적인 요인들에 의해 기인될 수 있으므로 주요 지배 인자에 대한 메커니즘적 이해라는 도전 과제가 남아있다.
2.4.2 바나듐 산화물계 음극 소재
바나듐 산화물의 경우, 5족의 바나듐 이온을 포함하는 산화물로, 타이타늄 다음으로 가벼운 원소를 포함하고 있다. 바나듐 산화물은 다양한 조성과 구조에서 V2+ 부터 V5+까지 폭넓은 산화수를 나타냄으로, 이온 저장 메커니즘에 대해 많이 연구되어왔다.[55] 이온 저장 호스트 재료로써 바나듐 산화물은 VO2, V2 O3, V2 O5, V2 O5⋅ nH2 O, V3 O7⋅ H2 O, V6 O13 등과 같이 다양한 화합물 형태로 존재하며 다양한 산화수를 갖음으로써 LIBs의 양극 재료부터 음극 재료로까지 연구되며 폭 넓은 반응 전위 영역을 보였다.[43] 그 중 상대적으로 낮은 V3+ 산화수를 가지는 V2 O3는 능면 정계(rhombohedral system) 결정질로 공간군(space group) R 3 ¯ c
바나듐 산화물 계열 중 음극 소재로 가능성을 보였던 VO2(vanadium dioxide)는 다수의 다형성을 지녔고, 각 상마다의 이온 저장 성능이 상이하였지만, bronze 구조의 VO2(B)에서 우수한 전기화학적 성능이 많이 확인되었다.[60] VO2(B) 상은 [VO6] 팔면체 이중층으로 구성된 층상 구조를 가지며, 각 이중층은 모서리를 공유함으로써 연결되어 층 사이에 이온 확산 경로가 1차원 채널로 존재한다. Yan 연구팀에서 보고한 SIBs용 음극 VO2(B)/ rGO의 경우 100 mA g-1의 전류밀도에서 약 400 mAh g-1에 가까운 가역 용량을 나타내었고, 4 A g-1의 고속 전류 밀도에서 약 200 mAh g-1의 초기 가역용량을 2000 싸이클동안 73.5% 유지하는 우수한 특성이 보고된 바 있다.[61]
2.5 인 기반 음극 소재 연구
위 2.4절까지 상대적으로 전극 부피 변화가 작은 삽입/탈리 반응을 보이는 음극들에 대한 내용이 다루어졌다. 삽입/탈리 반응 전극들의 안정적인 충-방전 거동은 LIBs 시스템의 상용화를 가능하게 하였으나, 근원적으로 낮은 이론 용량 특성으로 인해 고에너지밀도화에 많은 어려움이 있다. LIBs 음극에서 약 4200 mAh g-1의 가장 높은 이론 용량을 가지는 합금 반응 음극 실리콘이 SIBs에서 1/4 수준의 이론용량 밖에 가지지 못함으로써, 인(P, phosphorus) 원소가 SIBs에서 이론 용량이 2596 mAh g-1으로 가장 높은 소재가 되었으며, 산화/환원 반응 전위 구간이 약 0.3-0.7 V(vs. Na/Na+) 사이로 충분히 낮아 고에너지 밀도 음극 소재로 많은 이목을 집중 받아왔다.[62] Fig. 5.a 에 보이듯이 인은 여러 동소체(allotropy)를 가지고 있는데, 본 논문에서는 주요 3개의 동소체인 백린(white phosphorus), 적린(red phosphorus), 흑린(black phosphorus)에 대해 다루고자 한다.[63]
Fig. 5.
a. Crystal structures of three polymorphs of phosphorus(P) (white P, Red P, and Black P). Reprinted with permission.[63] Copyright 2014, American Chemical Society. b. Representative characteristics of three polymorphs of P (white P, Red P, and Black P). Reprinted with permission.[64] Copyright 2018, Wiley-VCH Verlag GmbH & Co. KGaA
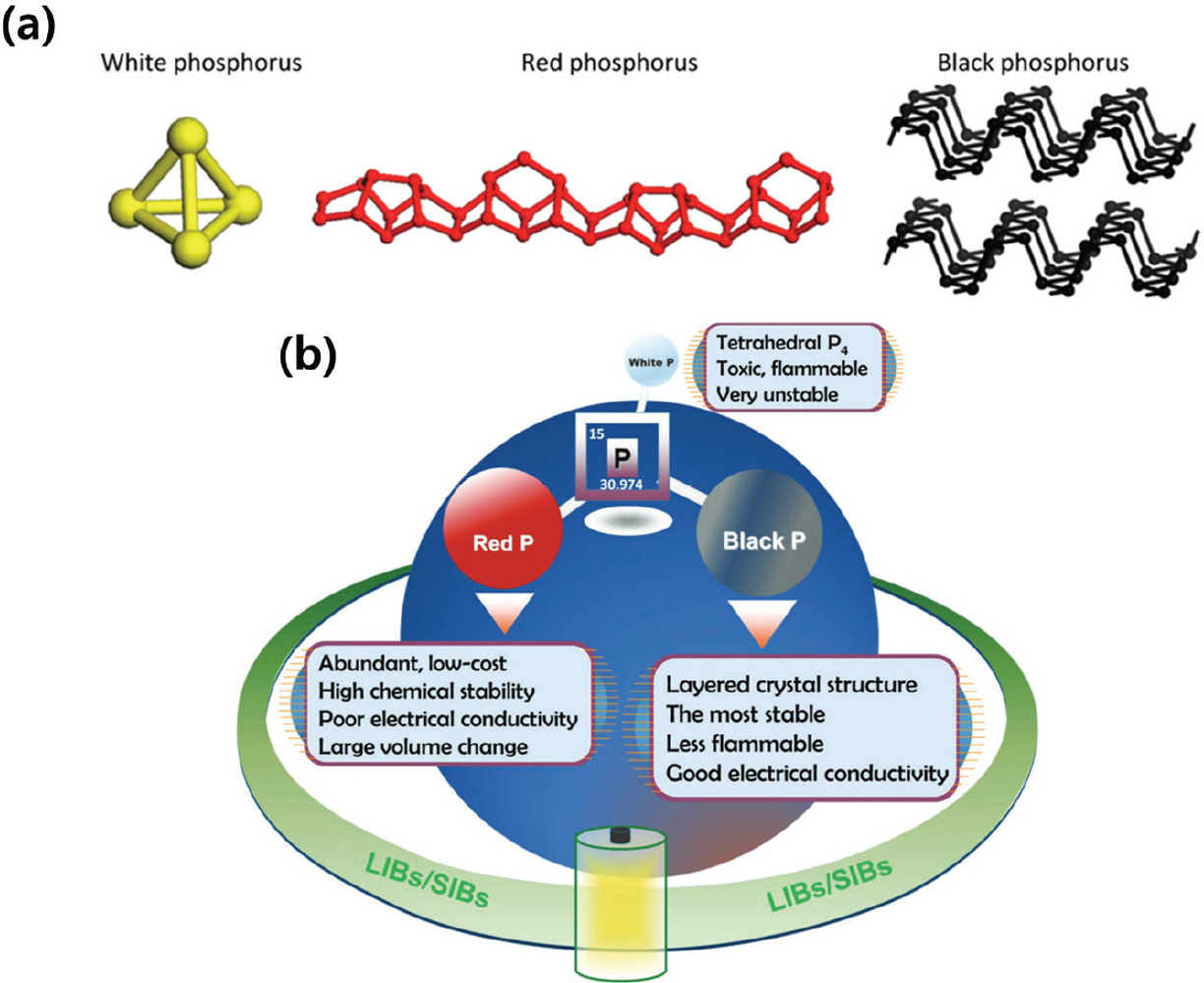
먼저, 백린은 사면체 구조(tetrahedral structure)의 P4 분자를 이루며 P-P 결합(약 200 kJ mol-1)을 하고 있다. 이 구조는 큰 결합 변형(bonding strain)으로 인해 화학적으로 불안정하여 다른 동소체로 쉽게 변할 수 있다. 약 40° C에서 자발적으로 연소되며 독성이 매우 강하기 때문에 전극재의 안전성과 성능 신뢰성에 큰 위협이 되어 연구 개발에 어려움이 있다(Fig. 5.b).[64] 반면, 적린의 경우, 백린을 260° C에서 열처리하여 얻을 수 있고 열 뿐만 아니라 빛 또는 X-ray로도 쉽게 상변태(phase deformation)가 가능하다. 적린은 주로 비정질 고분자 사슬 모양 구조를 형성(Fig. 5.a)하여 P4 결합의 분리와 화학적 결합에 의한 등변 삼각형(equivalent triangle)의 연결로 이루어져 있어 낮은 휘발성 및 높은 화학적 안정성을 부여한다. 이처럼 백린에 비해 적린은 매우 안정적이므로 음극 소재로 개발이 추진되었고 소듐 이온과 합금 반응을 통해 인 원자 하나당 저장 가능한 소듐 이온의 양이 3개로 많아 고용량 전극 소재로 적합하다. 마지막으로 인의 동소체로 소개하는 흑린은 열역학적으로 동소체 중 가장 안정하여 대부분의 용매에 용해되지 않고 불연성의 특성을 가지며, 층상 구조의 주름진 시트 형태(Fig. 5.a)로 존재한다. 흑린은 흑연과 비슷한 층상구조를 가지지만, 내부 층의 큰 채널이 소듐 이온을 충분히 받아들여 안정적인 이온 출입이 가능하고 흑린은 우수한 전기 전도성이 있어 음극 소재의 성능적인 면이 우수하다. 그러나, 흑린 합성을 위해서는 매우 높은 압력과 온도를 필요로 하여 합성 및 제조 측면에서 제한되는 점이 많아 현재로는 실제 음극 소재로 활용되기에는 경제적 측면의 어려움이 따른다.[65]
전극 소재로 실용 가능성이 보이는 인의 소듐화 과정은 P → Na x P → NaP → Na2 P → Na3 P순의 단계별 합금화 방식으로 이루지며 큰 부피 변화에 의한 전극 구조 불안정성을 막기 위해 인-카본 복합체(phosphorus-carbon composites) 제조에 대한 연구가 많이 이루어져왔다.[66,67] 적린은 충전 과정 중 300∼500% 이상 입자의 큰 부피 변화가 발생하여 전극 단위에서의 박리와 입자 균열 및 파쇄로 음극에서의 전기화학적 성능이 급격하게 악화될 수밖에 없다.[68] 게다가 적린의 낮은 전기전도성(≈10-12 S m-1)은 충-방전 반응 시 전극 내 저항을 크게 증가시키는 요소로 작용하는 문제가 있다.[64,68]
이러한 인의 낮은 전기전도성과 수명 특성을 보완하기 위해 다양한 탄소 기반 물질을 함께 복합체로 형성하여 전기 전도성을 증가시키는 연구가 다수 이루어졌다.[69-72] Sun의 연구팀에 의하여 연구된 인의 LIBs 용 음극 연구에 의하면, 적린의 낮은 전기 전도로 인해 초기 충전 용량이 약 1700 mAh g-1이었으나, 방전 시 약 200 mAh g-1 으로 적린의 낮은 전기 전도로 인해 매우 큰 용량 손실이 나타났다.[63] 하지만, 기계화학적 합성법(mechanochemical synthesis) 중 하나인 고에너지 밀링 (HEMM, high energy milling method)을 통하여 흑린과 흑연을 잘 혼합하는 경우, 적린에 비해 높은 흑린의 전기전도성에 기반하여 0.2 C 기준, 초기 약 2250 mAh g-1의 가역 용량을 시작으로, 100 싸이클 이후 1849 mAh g-1의 높은 가역용량을 유지하는 우수한 특성 거동이 관찰되었다.[63] 이로써 인은 고용량 음극 소재로 가능성이 확인되었으나, 적린의 낮은 전기 전도성과 합금 반응 메커니즘에 의해 생기는 충-방전 과정 급격한 부피 변화로 인해 저조한 수명 특성을 드러냈으며, 전기적 특성을 보완하는 도전재와 구조적 안정성을 증가시키기 위한 첨가제 등에 대한 후속 연구가 진행되고 있다.
2.6 금속 인화물 기반 음극 소재 연구
금속 인화물(metal phosphides)은 인과 금속 양이온이 결합한 화합물 형태로 적린의 가연성, 낮은 전기전도성, 및 큰 부피 변화 문제를 해결해고자 연구되고 있는 음극 물질이다. 최근 몇 년 간 인의 장점인 고용량 특성을 나타내면서 인의 단점을 보완한 인 기반의 화합물로서 금속 인화물이 LIBs 및 SIBs용 음극분야에서 큰 주목을 받아왔다. 금속 인화물의 리튬 또는 소듐 이온과의 반응 전위가 앞서 소개한 금속 산화물 또는 금속 황화물 등 다른 화합물에 비해 훨씬 낮은 특징을 보이기 때문에, 고용량 특성과 더불어 고에너지 밀도 소재로 적합하다고 할 수 있다.[72] 금속 인화물 음극 소재군은 앞서 소개한 금속 산화물 소재와 마찬가지로 화합물을 구성하는 금속 양이온의 종류에 따라서 리튬 이온 저장 메커니즘이 달라지는 것이 확인되어졌다.[67,72] 인화물을 구성하는 전이금속과 인 간의 화학양론 비율이 1:1 인 일인화물(mono-phosphide)을 기준으로 보면, 초기 전이금속인 Ti로 구성된 TiP와 V로 구성된 VP 상, 그리고 Mo로 구성된 MoP의 경우는 전이 금속의 큰 이온 반경으로 인해, 리튬 이온이 삽입되기 충분한 크기의 프리즘 자리(prismatic site)를 형성할 수 있다. 또한, 초기 전이금속의 전기 음성도가 충분히 낮아 Li/Li+ 환원 전위 근처에서도 전이 금속이 환원되지 않고 결정 구조를 유지하여 삽입되는 반응으로 리튬 이온을 저장한다. 반면, Ti과 V 다음에 존재하는 Mn으로 구성된 MnP 구조의 경우, Li/Li+ 환원 전위 근처에서 Mn 이온이 금속으로 완전히 환원되는 것이 아닌 합금화 반응으로 리튬 이온을 저장하게 되며, 후기 전이금속(later transition metal)인 Fe, Co, Ni로 구성된 FeP, CoP, NiP 음극들은 Li/Li+ 환원 전위 근처에서 각 전이금속 양이온들이 완전히 금속 상태로 환원되면서 P 음이온이 리튬 이온과 반응되는 전환 반응 메커니즘(TMP + 3 Li+ + 3 e- → TM 0 + Li3 P,TM = transition metal)을 보이게 된다.[67] 각 이온 저장 메커니즘에 따라 전기화학적으로 고유한 장/단점을 가지게 됨으로 해당 조성들을 초기 및 후기 전이 금속 인화물로 나누어 그에 대한 연구 동향을 소개하고자 한다.
2.6.1 초기 전이금속 인화물
상기 소개한 초기 전이금속 인화물의 대표적인 조성은 TiP, V-P, 및 MoP 시스템으로 다양한 조성에 대해 LIBs용 음극으로 연구되었고 유망한 조성에 대해 SIBs용 음극 연구로 확장되어가는 추세이다. 먼저, 타이타늄 기반 인화물(TiP, TiP2, TiP4)은 V-P 및 MoP 시스템 대비 상대적으로 음극 소재 측면의 연구가 많이 이루어지지 않았다. Woo 연구팀에 의하면, 다양한 조성비의 Ti/P(1/1, 1/2, 1/4) 원료 분말을 이용, 고에너지 밀링으로 만들어진 TiP 복합체가 LIBs용 음극으로 연구되었다.[73] Ti/P 비율이 1/1로 합성된 복합체는 Ti4 P3– Ti5 P3– P로 유의미한 가역 용량이 확인되지 않았고, 1/2 비율의 경우 TiP– Ti4 P3– Ti5 P3– P 복합체로 첫 싸이클에서 831 mAh g-1의 비교적 높은 용량을 나타냈으나, 이후 싸이클에서 빠르게 감소하는 거동을 보였다. 그에 반해, Ti/P 몰 비율이 1/4인 경우 P-TiP2 복합체를 형성했고 리튬 이온과 반응 시 Li10.5 TiP4 상으로 변하는 합금 반응을 나타내며 초기, 1420 mAh g-1의 높은 가역용량을 나타내었으나, 5번째 싸이클에서 가역 용량이 400 mAh g-1으로 빠르게 감소하였다.[73] 이처럼, 현재 TiP 관련 소재들은 여러 상들이 혼재된 복합체에 대해서 주로 연구가 이루어져 있어, 각 조성 상의 명확한 이온 저장 메커니즘이 밝혀지지 않았고 각 상들이 SIBs 용 음극에 적용된 사례도 상당히 적다고 할 수 있다.
반면에, 다양한 조성(V3 P, V2 P, V12 P7, V4 P3, VP, V4 P7, VP2, VP4)의 V-P 시스템에서는 VP, V4 P7, VP2, VP4 등 다수 조성에 대해 LIBs용 음극으로 연구가 되었으며, 조성 순으로 삽입, 합금, 합금, 및 전환 반응으로 이온 저장 메커니즘이 달라지는 거동을 보였다.[74-78] 특히, VP 상의 경우 hexagonal 구조를 가지면서 구조 내 존재하는 프리즘 자리에 리튬 이온이 가역적으로 삽입/탈리되어 약 250 mAh g-1의 용량을 장수명 유지하는 특성으로 인해(Fig. 6.a)[74] SIBs용 음극 소재로의 도입이 기대되었지만, 예비적인 실험 결과로는 흑연의 경우와 유사하게, 소듐 이온의 삽입이 일어나지 않는 것으로 보여진다. 하지만, Kim 연구팀에 의해 VP와 조금 다른 화학양론의 V4 P7(V:P = 1:1.75) 조성에서 결정 구조의 변화 없이 소듐 이온의 삽입/탈리(V4 P7 + xNa+ + xe- ↔ Na x V4 P7)가 가능함이 XRD, TEM, XANES 등의 분석을 통해 메커니즘적으로 밝혀졌다. 합성된 V4 P7 상은 2.4.2 절에서 소개된 바나듐 산화물 계열의 유망한 후보군 VO2(B)와 비교 분석되어 졌는데, VO2(B)의 가역용량보다 조금 높은 250 mAh g-1의 가역용량을 보이면서, 반응 전위 역시 상대적으로 낮아 산화물 음극재에 비해 높은 에너지 밀도 특성을 보여주었다(Fig. 6. b, c). 특히, V4 P7 상을 카본 소재와 복합체를 형성하지 않더라도, 단일상으로도 약 122 mAh g-1의 가역용량(전류 밀도 500 mA g-1)을 500 싸이클동안 96% 유지(Fig. 6.d)하는 것이 확인되었다.[79] 이러한 V4 P7 상의 안정적인 거동으로부터, 추가적인 용량 향상을 유도하기 위해, Kaushik 연구팀이 HEMM을 통해 V4 P7/5P 복합체를 합성하였지만, 잔존하는 인(P)이 Na3 P를 형성하는 합금 반응 시 생기는 급격한 부피 변화로 인해 전극의 안정성이 떨어져 수명 특성이 크게 감소하였으며[80], V4 P7/P 간의 비율 최적화 등의 추가 연구가 필요해 보인다.
Fig. 6.
a. Proposed topotactic insertion reaction mechanism of VP (V atoms, green; P atoms, purple; Li atoms, red). Reprinted with permission.[74] Copyright 2009, American Chemical Society. Galvanostatic discharge/charge voltage profiles for b. V4 P7/Na and c. VO2(B)/Na cells tested at a current density of 50 mA g-1. d. Cycling performance of V4 P7/Na and VO2(B)/Na cells at a current density of 500 mA g-1 with an activation of each 5 cycles at 50, 100, and 200 mA g-1. Reprinted with permission.[79] Copyright 2019, Royal Society of Chemistry

2.6.2 후기 전이금속 인화물
후기 전이금속으로 이루어진 인화물의 경우 MnP, FeP, CoP, NiP, Cu-P 등 다양한 조성에 대해 LIBs 용 음극소재로 연구되었으며, 대다수 조성에 대해 SIBs 용 음극소재로도 연구가 많이 이루어진 물질군이라 할 수 있다.[72,81] 후기 전이금속 인화물은 LIBs용 음극으로 적용하여 Li/Li+ 환원 전위까지 반응 시, 합금 반응을 보이는 MnP 구조를 제외하고는 전이금속/P의 비율 및 후기 전이금속 종류에 상관없이 금속 양이온과 P 사이 결합이 끊어지면서 양이온이 금속으로 환원되는 전환 반응 메커니즘을 보였다.[82,83] 때문에, 후기 전이금속 인화물 내 인의 함량이 높은 상(P-rich phase)일수록 이론 용량이 커지지만 그에 따른 충-방전 시 부피변화율이 커져 안정성이 떨어지는 상충관계를 보이게 된다.
먼저 소개할 MnP 계열 내 가장 인 함량이 높은 MnP4는 팔면체 구조(octahedral structure)의 MnP6 배열에 따라 2-MnP4, 6-MnP4, 8-MnP4, γ-MnP4의 동질 다형체(polymorph)로 분류된다. Kim 연구팀은 HEMM법으로 삼사정계(triclinic)구조를 가진 6-MnP4(Fig. 7.a)를 처음 합성하여 LIBs 및 SIBs 음극소재로 도입하였다.[84] LIBs용 음극으로 적용 시, MnP4 + 7Li+ + 7e− → Li7 MnP4의 합금반응 이후 Li7 MnP4 + 5Li+ + 5e− → Mn0 + 4Li3 P의 전환 반응 메커니즘으로 발현되는 높은 초기 충전 용량(1876 mAh g-1)과 가역 용량(1615 mAh g-1)을 보였다(Fig. 7.b). SIBs용 음극으로 활용할 때는 중간에 형성된 합금 반응상 없이 곧바로 전환 반응이 진행되었고, 1234 mAh g-1 및 1028 mAh g-1의 높은 초기 충전 및 가역 용량이 확인되었다(Fig. 7.c). 추가적으로 연구 결과, 합성된 MnP4와 graphene nanosheet 의 복합체 형성을 통해 MnP4 분말의 큰 부피 변화와 응집을 완화하여 718 mAh g-1의 높은 초기 용량을 100 싸이클동안 87.3% 유지하는 연구 결과가 보고되었다.[84]
Fig. 7.
a. Crystal structure illustration and Galvanostatic discharge/charge profile and ex-situ XRD patterns of triclinic 6-MnP4 for b. Li half cell and c. Na half cell, respectively. Reprinted with permission.[84] Copyright 2021, Wiley-VCH Verlag GmbH & Co. KGaA. d. SEM image and e. cycling performance of as-prepared CoP4/carbon fiber electrode tested at a current density of 300 mA g-1 in Na half cell. Reprinted with permission.[86] Copyright 2018, Wiley-VCH Verlag GmbH & Co. KGaA

CoP 계열의 경우 다른 전이 금속에 비해 코발트 원료 값이 비쌈에도 불구하고 Co2 P부터 CoP, CoP2, CoP3, CoP4 등 다양한 조성에 대해 LIBs용 음극 소재로 도입 및 연구가 되었으며 위 명시된 조성에 대해 모두 SIBs용 음극으로 연구가 수행되었다.[85,86] 그 중 P-rich한 CoP3 상의 경우, LIBs 음극으로 보였던 높은 용량 특성이 구현되지 않고, CoP3@C 복합체에서 전류 밀도 100 mA g-1 기준, 약 300 mAh g-1의 낮은 가역용량만 구현되었다.[87] 반면, CoP4상의 경우, 탄소 펠트(Carbon felt) 위에 나노 구조 형태의 배열을 가지게 합성되어 초기 충전 용량은 약 1690 mAh g-1로 높게 나타났고, 2 싸이클부터에서 약 900 mAh g-1만큼의 높은 가역 용량을 나타내었다(Fig. 7.d, e). 충-방전 시 CoP4 상의 큰 부피 변화가 예상됨에도 불구하고, 초기 싸이클에서 용량이 증가하는 활성화 과정이 발생한 이후 300회 싸이클 후에도 851 mAh g-1의 가역 용량을 유지하는 우수한 장수명 특성을 보였다.[86]
다음은 원료 값이 저렴하여 유망한 후보군이 될 수 있는 FeP 계열의 인화물을 소개한다. FeP 계열에서도 FeP, FeP2, FeP4 등에 대해 LIBs용 음극 소재로 연구 되었으며 위 조성들도 모두 SIBs용 음극으로 연구가 수행되었다.[88,89] Zhang 연구팀은 HEMM법으로 합성된 FeP2와 FeP4에 대하여 SIBs용 음극 특성을 보고하였는데, 합성된 FeP2는 Pnma 공간군을 가지며 사방정계 결정구조를 가졌고, Fe 원자 주변 왜곡된 FeP6 팔면체가 위치한다.[90] 이들은 가장자리를 공유하여 팔면체 체인을 형성하여 모서리에 서로 연결되는 반면, FeP4의 경우 P21/c의 공간 군과 단사정계 결정 구조를 갖고 FeP4 결정 구조에 가장자리를 공유하는 FeP6 팔면체로 이루어진 팔면 삼합체(octahedron trimer)가 포함되어 있다. FeP2와 FeP4와의 소듐 이온 저장 거동을 분석한 결과, FeP2는 소듐 이온과의 반응에서 유의미한 전기화학적 활성이 나타나지 않았다.[90] 그에 반해, FeP4는 첫 소듐화와 탈소듐화 시 각각 1417과 1145 mAh g-1의 용량으로 초기 쿨롱 효율이 약 81%로 나타났으며, 소듐 이온 저장 메커니즘이 Na3 P 상을 형성하는 전환 반응임이 확인되었다.[90] 관찰된 수명 특성 역시, 약 1100 mAh g-1의 가역 용량을 30 싸이클 유지하는 등, 유망한 거동을 보여, 입자 사이즈 조절 및 카본 복합체 형성 등의 추가 특성 연구가 기대되어지는 물질이라 할 수 있겠다.
대표적인 후기 전이금속 인화물의 마지막 예시로, NiP계 인화물의 경우 Ni3 P, Ni5 P2, Ni12 P5, Ni2 P, Ni5 P4의 Ni-rich한 상에서 NiP, NiP2, NiP3와 같은 P-rich한 상이 존재하며, Ni2 P, NiP2, NiP3 상 위주로 SIBs용 음극 연구가 이루어졌다.[83,91] Ni2 P는 P-rich한 NiP 상들에 비해 합성 반응 속도가 빠르고 mild한 조건에서 합성이 가능하여 다양한 나노 구조 형태가 적용되어 연구되었다. Wang 연구팀은 Ni2 P 나노 시트 구조를 카본 천 위에 성장시킴으로써 100 싸이클 이후 가역용량 399 mAh g-1를 유지하는 효율적인 3D 구조의 음극 소재를 개발하는 등 많은 연구가 이루어지고 있으나,[72] 화합물 조성에서 오는 이론 용량의 한계(낮은 인 함량)로 인해 고용량 특성을 기대하기는 힘들다. 반면, NiP2의 경우 NiP2 + 6Na++ 6e− → Na3 P + Ni0의 전환 반응을 통해 약 1333 mAh g-1의 이론 용량을 나타낼 수 있다. 하지만, Kim 연구팀에 의해 HEMM법을 통하여 합성된 나노 NiP2 분말은 전류 밀도 50 mA g-1에서의 초기 충전, 방전 용량이 각각 653 mAh g-1, 489 mAh g-1로 초기 쿨롱 효율이 75%로 이론 용량에 크게 못 미치는 특성이 관찰되었다. 이는 반응 전위가 낮아 고전압 구현이 가능했던 인화물의 장점이 오히려 소듐 이온 확산 시 발생하는 큰 분극(polarization)에 의해 주어진 반응 전위 영역 내에서 인화물이 소듐 이온과 충분히 반응하지 못하는 현상으로 해석할 수 있다.[92] 이에 대한 해결방안 중 한가지로, 다음 2.7절에서 음이온 치환형 고용체 Ni2-x S x 소재에 대해 소개하도록 하겠다. NiP3상은, NiP 계열에서 가장 P-rich한 화합물로 NiP3 + 9Na+ + 9e− → 3Na3 P + Ni의 전환 반응 메커니즘을 가진다. NiP3에 관하여 보고된 연구는 HEMM으로 합성된 NiP3 나노 분말에 카본 나노 튜브(CNT)를 복합체화하여 그 특성을 보고하였다.[93] NiP3/CNT 복합체는 전류밀도 100 mA g-1 기준, 868.4 mAh g-1의 높은 가역 용량을 나타내었으며, 약 1.6 A g-1의 고속 충-방전에서 200 싸이클 동안 65%의 용량을 유지하였다.[93]
2.7 금속 황화물 기반 음극 소재 연구
금속 황화물(MSs, Metal sulfides)은 금속 양이온과 16족 음이온(S, Se, Te 등)으로 구성된 금속 칼코겐 화합물(calcogenides) 중 한 가지로 앞서 소개된 금속 산화물과 인화물에 비해 상대적으로 높은 이온 저장 반응 전위를 나타낸다. 이 때문에, 초기 전이 금속으로 구성된 화합물에서 삽입/탈리 반응을 보였던 산화물(e.g., TiO2와 VO2 등), 인화물(TiP, VP, 그리고 MoP 등)과는 달리 금속 황화물에서는 초기 전이 금속으로 이루어진 TiS, V-S, 및 MoS 계열에서도 Li/Li+ 또는 Na/Na+ 환원 전위 근처에서 금속 양이온이 모두 환원되는 전환 반응을 보이게 된다.[94,95] 이 때문에, 전이금속의 종류 및 금속/황 간의 비율에 관계없이 대다수가 전환 반응(e.g., MS2 + (4 – x)Na+ + (4 -x)e− ↔ 2Na2 S + M) 메커니즘을 나타내고 있다. 특히, 이차원 층상 구조를 가지는 MS2 구조의 TiS2, VS2, MoS2, 등에 대한 연구 보고가 다수를 이루고 있고[96] 해당 구조들은 반응 초기, MS2 구조 내의 침입형 자리에 리튬 또는 소듐 이온이 삽입된 이후 반응 전위가 더 낮아짐에 따라 전환 반응이 일어나게 되는 형태이다. 금속 황화물이 SIBs용 고성능 음극으로 적용되기 위해서 넘어야 할 가장 큰 장벽은 산화물 또는 인화물에 비해 높은 반응 전위이다.[97] 금속 황화물은 반응전위가 상당히 높기 때문에 앞서 인화물 음극재의 문제로 다루어졌던, 소듐 이온 확산 시 발생하는 큰 분극으로 인해 충분하지 못한 반응 전위와 같은 문제는 없으며, 오히려 대다수의 금속 황화물이 충분히 반응하여 우수한 용량 특성이 관찰된다. 하지만, 충-방전에 사용되는 용량이 구현되는 전압이 상당히 높기 때문에 양극과 풀 셀을 형성하였을 때 전지 전체의 에너지 밀도 측면에서 불리하게 작용한다.
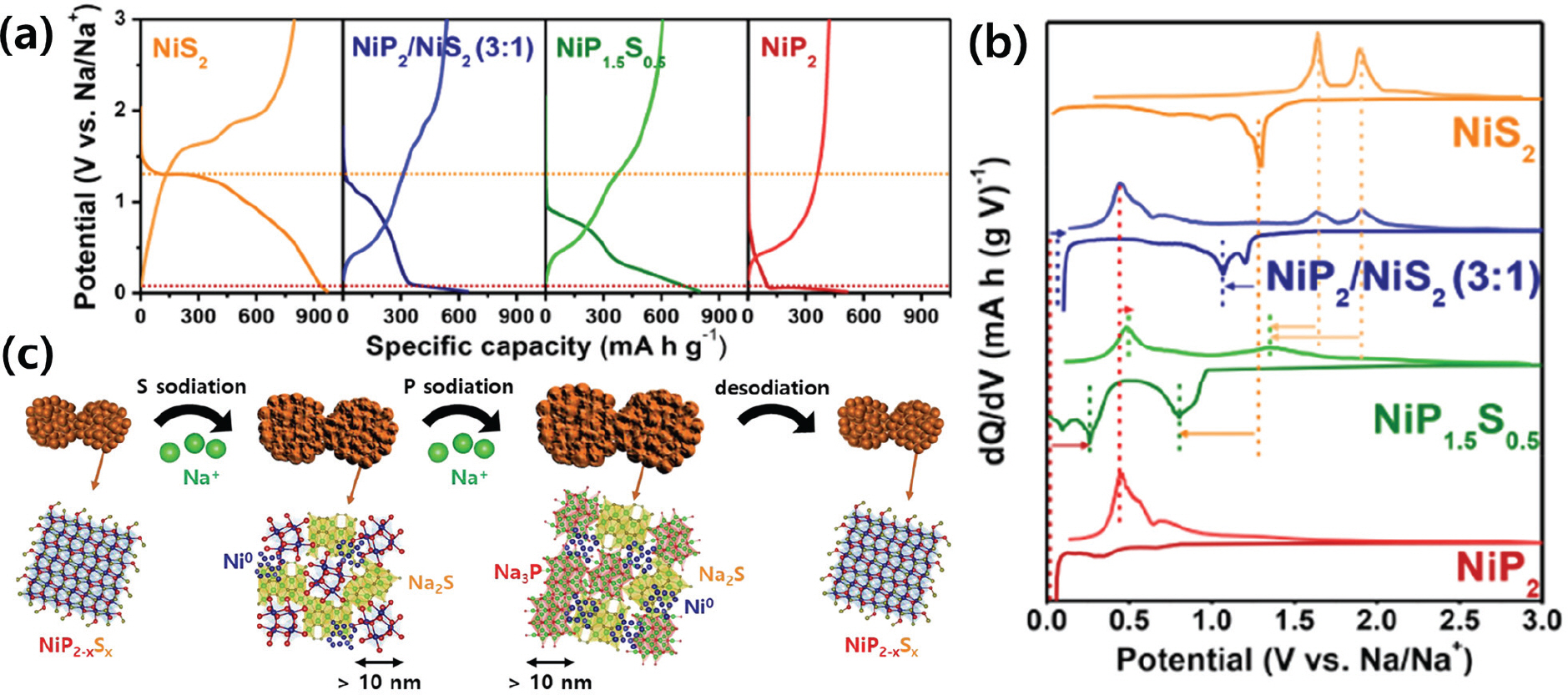
이러한 금속 황화물의 높은 반응 전위를 개선하기 위한 한 가지 접근법으로, 최근 음이온 치환을 통한 산화/환원 전위를 열역학적으로 변화를 주는 접근이 시도되었다. Kim 연구팀에 따르면, 높은 반응 전위를 가진 금속 황화물 NiS2 상과 낮은 반응 전위를 가지면서 NiS2와 동일한 결정 구조를 가지는 NiP2 금속 인화물을 음이온 치환형 고용체 NiP2-x S x로 형성함으로써 각 인과 황의 전환 반응 전위가 크게 변화되는 결과를 보고하였다(Fig. 8.a, b).[98] 이들은 NiP2-x S x를 HEMM법을 통해 전 조성 범위 내 음이온 치환 고용체를 성공적으로 합성하여 각 인과 황의 반응 전위를 큰 폭으로 변화시켰다. 이를 통해, 너무 낮아 문제가 되었던 금속 인화물의 반응 전위를 높이는 동시에, 너무 높은 금속 황화물의 반응 전위를 감소(Fig. 8.b)시키면서, 가역 용량 및 전압의 최적화를 NiP2-x S x (x=0.25) 조성에서 달성하였다. 또한, 치환형 고용체의 경우 원자 단위에 가까운 형태로 인과 황의 음이온이 섞여 있기 때문에, 전환 반응을 통해 형성된 Ni/Na3 P/Na2 S 매트릭스가 나노 복합체를 형성하여, 충-방전 과정 중 음극 소재의 체적 변화를 완화하고 높은 이온 확산 속도를 제공하는 결과(Fig. 8.c)를 보였다. 이처럼 새로운 이성분계 조성의 금속 화합물에 대한 연구 이외에도, 삼성분계 또는 사성분계로 조성의 범위를 넓히거나, 또는 소재의 결정학과 이온 저장 메커니즘 간 관계를 활용해 디자인한 양이온 또는 음이온 치환형 고용체 도입 등으로 향상된 전기화학적 성능을 이끌어 낼 수 있는 연구들이 최근 시도되면서 많은 주목을 받고 있다.[84,98-100]
Fig. 8.
a. The 1 st cycle discharge/charge voltage profiles, and b. corresponding dQ/dV plots at a current density of 50 mA g-1 of the NiP2-x S x (x=0, 0.5, and 2.0) and NiP2/NiS2 (3:1) electrodes. c. Schematic illustration for the sodiation/desodiation process of the NiP2-x S x solid solution. Reprinted with permission.[98] Copyright 2023, Elsevier
결론
본 논문에서는 차세대 소듐 이온 전지용 고성능 음극 소재를 위한 연구 개발 동향을 소개하며 현재 연구 개발되고 있는 주요 음극 재료들과 각 조성의 재료들이 갖고 있는 열역학-결정학-화학적 관계에 의해 결정되는 이온 저장 메커니즘과 전기화학 성능을 소개하였다. 소듐 이온의 경우 리튬 이온과 비교하여, 경제적인 측면에서 원료가 저렴한 장점이 있지만 이온 반경이 상대적으로 크고 무겁기 때문에 에너지 밀도가 낮으며 이온의 확산 및 이동에 있어 저항이 큰 단점들이 존재한다. 또한, 기존의 리튬 이온 전지에 적용되었던 음극 소재들의 경우, 소듐 이온 전지에 적용되었을 때 다른 이온 저장 메커니즘을 보이는 경우가 관찰되는데, 이는 리튬에 비해 소듐의 표준환원 전위가 높고, 소듐의 이온 반경이 더 크기 때문에 음극 소재 내 저장되는 메커니즘이 달라질 수 있는 것에 기인한다. 특정한 음극재의 리튬 이온 저장 메커니즘과 소듐 이온 저장 메커니즘이 유사하더라도, 소듐 이온 저장 시 발생하는 부피 변화율은 기존의 리튬 이온 저장 시의 부피 변화율과 다르기 때문에, 그에 적합한 복합체, 고용체, 및 전극의 디자인이 이루어져야 한다. 본 논문에서 소개된 각 조성 소재들의 이온 저장 성능은 전기화학적 이온 저장 메커니즘으로부터 기인되는 것임으로, 리튬과 소듐의 화학 및 전기화학적 성질의 차이를 잘 이해하고 이것이 이온 저장 메커니즘과 어떻게 연관이 되는지를 잘 이해하는 과정이 고성능 음극 소재를 디자인하는데 주요한 정보를 제공한다고 할 수 있다. 본 논문에 소개된 다양한 소재들과 연구 전략들로부터 파생된 형태의 심화 연구가 더해진다면, 리튬 이온 전지와 더불어 경제성이 우수한 차세대 소듐 이온 전지의 상용화가 더욱 앞당겨 질 것으로 기대한다.






